
前言
在上一篇关于药品GMP比较法研究的文章《一文读懂中美欧GMP监管差异|GMP比较法研究(上)》中,我们从监管法律体系、监管机构及人员以及GMP检查程序三个方面对我国与美国、欧盟的药品GMP监管进行了比较。正因为各国监管机构的GMP监管标准及程序存在差别,在涉及药品进出口及跨境委托生产时,如何就该等药品开展GMP监管成为各国药品监管棘手的难题,也成为开展药品进出口及跨境委托、受托生产业务的药品企业最为关心的问题之一。前段时间,我国国家药监局叫停美国Celgene Corporation注射用紫杉醇(白蛋白结合型)进口并取消其集中采购中选资格,就是国家药监局在执行对从境外进口药品的监管职能,其依据为我国的GMP监管标准。
本文将从国际上通行的GMP监管互认制度出发,介绍美国、欧盟及我国目前的药品GMP监管互认状态,并基于此谈谈美国FDA和我国国家药监局是如何开展GMP跨国监管的。
01 药品GMP监管互认概述
基于对药品质量的把控及监管要求,各国的药品监管机构通常会对于以下两类境外生产药品(“境外生产药品”)的生产企业按照本国的GMP标准进行现场检查:①进口药品的生产企业,以及②境内药品的境外受托生产企业,并根据检查结果决定是否允许受检药品进口、上市或销售,从而保证本国公民的用药安全。但因跨境监管耗时耗力,有限的监管资源越来越难满足各国对境外生产药品的跨境检查需求,在各国家/地域的药品监管机构之间实行GMP监管互认,能够在保证药品安全的同时促进合规标准的一致性发展,避免了不同国家/地域的药品监管机构对药品生产的重复性检查工作,节省了监管资源,同时也降低了药企应对重复性检查的成本。
药品GMP监管互认存在以下几种主要方式:
1.1 签署互认协议
各国家/地域的药品监管机构基于对其他国家/地域药品监管能力的评估和认可签署互认协议(Mutual Recognition Agreements,“MRA”),并就互认范围、适用于现场检查豁免或必须进行现场检查的品种,以及互认程序、信息共享、药物警戒机制等双方关注的问题予以约定,以实现一方监管机构对另一方监管机构GMP检查结果的承认。但是,MRA的签署并不意味着一方监管机构一定不会重复对方监管机构已执行过的检查,签署双方的监管机构通常都留有在例外情况下对另一方管辖范围内的药品生产企业执行GMP检查的权利。[1]
1.2 加入PIC/S
国际药品监查合作计划(The Pharmaceutical Inspection Co-operation Scheme, “PIC/S”)成立于1995年11月,是世界上唯一由各国GMP检查权责机关组成的国际合作组织,其成立的目的在于促进GMP的国际合作及标准一致化。PIC/S是一个国际药品生产GMP互认组织,各成员国在自愿的基础上相互承认官方GMP检查;与MRA不同的是,PIC/S搭建的是一个非约束性、非正式的合作平台[2],而MRA对于签署双方具有法律约束力。该组织的任务包括:促进各国卫生主管机关的检查互信,制定统一的GMP规范,统一GMP认证系统,对检查认证人员统一培训(如即将于2020年11月在泰国曼谷举办的“如何成为优秀的GMP检查员”研讨会),促进GMP信息的交流。
为此,PIC/S制定了GMP检查互信指南,概述了境外药品生产企业GMP合规性的非现场评估过程,并确定在何时可通过其他或当地监管机构的监管动作来确认并保证企业处于可接受的GMP合规水平范围内,从而替代现场检查,避免了监管机构间的重复工作。[3]
PIC/S目前共有50余个监管机构成员,遍布澳大利亚、加拿大、捷克、丹麦、芬兰、法国、德国、新加坡、日本、英国、美国、台湾和香港等国家/地区。[4]监管机构成员均需要满足并维持PIC/S GMP标准,以作为其GMP检查互认的基础,该组织颁发的GMP证书在PIC/S组织成员之间可以相互认可,所以加入PIC/S是进入国际市场的快捷通道之一。
1.3 其他合作项目
除上述两种方式外,各国家、国际组织间还会不定期就特定项目开展合作,如2008-2016年的原料药检查国际合作项目,以及2019年12月刚刚启动且将持续至少2年时间的无菌药品GMP检查国际合作项目(“无菌药品合作项目”),参与方有FDA、EMA、法国药品保健品署(ANSM)、英国药品和健康产品管理局(MHRA)、澳大利亚药品管理局(TGA)、加拿大卫生部(HC)、日本药品医疗器械综合机构(PMDA)及世界卫生组织(WHO)。无菌药品合作项目的范围为人用化学无菌药品,以及某些治疗性生物制品(如单克隆抗体、重组蛋白),不含疫苗、细胞和基因疗法以及血液制品;根据项目工作计划,参与机构将协调和共享检查计划,并鼓励各方根据各自确定的GMP规范开展联合检查(避免独立或同步检查),每月共享检查结果。[5]
02 药品GMP监管的互认现状
2.1 欧盟对其他国家/地区GMP监管的承认
截至目前,欧盟已分别与澳大利亚、加拿大、以色列、日本、新西兰、瑞士及美国共7个国家签署了MRA,以在欧盟与该等国家之间实现药品GMP监管互认,以及就监管及质量问题进行信息共享等目的,但欧盟与各国签署的MRA范围均不相同。
在欧盟已签署的MRA中,最引人关注的要数与美国FDA签署的MRA。
这份MRA项下的互认范围为:
①双方在其领土范围内对已上市产品执行的生产检查;
②双方在其领土范围内对产品执行的上市前生产检查;
③双方在其领土范围外对产品执行的生产检查。
所覆盖的产品类型包括人用药品和生物制品(具体产品类别可在MRA的附录中找到);此外,欧盟和FDA应于2017年12月15日前决定是否将兽药包含在MRA所覆盖的产品类型中(根据FDA于2020年3月13日发布的公告,FDA兽药中心已于2019年12月向EC报告将兽药作为MRA所覆盖的产品,且欧盟已基本同意[6]),并于2022年7月15日前决定是否将人用疫苗和源于血浆的产品包含在MRA所覆盖的产品类型中;人血、人类血浆、人类组织和器官,以及兽用免疫物质不在MRA所覆盖的产品类型中。此外,值得关注的是,欧盟和FDA的MRA中也约定了一方拒绝认可另一方GMP检查结果的特殊情形,即在检查报告中存在原料不符或不足的迹象,在上市后监管中发现产品存在质量缺陷,或存在与产品质量或消费者安全相关的其他特殊证据等特殊情况下,欧盟和FDA可选择不接受对方就其领土范围内的生产企业所发布的官方GMP文件。[7][8]
欧盟负责MRA起草和谈判工作的是欧盟委员会(EC),EMA则负责向EC提供咨询服务,并在MRA签署后负责后续的执行工作,包括作为互认监管机构与欧盟成员国监管信息的对接口,负责就已签署的MRA进行答疑和管理,管理数据库,并与成员国的监管机构进行数据共享等。
2.2 美国对其他国家/地区GMP监管的承认
2012年7月9日,美国国会通过《食品、药品管理安全与创新法案》(Food and Drug Administration Safety and Innovation Act),其中规定,如果FDA认为其他国家/地区的监管机构能够达到美国监管标准,则FDA有权与该境外监管机构对该监管机构境内药品生产的监管进行互认。[9]但美国的药品GMP互认门槛很高且FDA对互认非常谨慎,根据FDA官网显示,美国截至目前仅与欧盟签署了MRA,且FDA虽然在2017年7月与欧盟签署了MRA,但因对欧盟各成员国GMP监管机构的检查工作质量持保守态度,因此并没有在MRA生效时就对所有欧盟成员国的GMP监管进行承认,而是约定了过渡期,并在过渡期内比照美国的现场审查标准对欧盟成员国的生产检查稳定性逐个进行审查,历时两年的时间才完成了对欧盟28个国家GMP监管的承认。
2.3 我国与其他国家/地区GMP监管体系的互认
2017年7月,国际人用药品注册技术协调会(ICH)确认中国药监局成为ICH第8个监管机构成员,使得中国药品在研发和注册领域的国际化道路上迈出了重要一步,但在药品生产监管领域,中国的监管体系还未加入PIC/S,中国也尚未与任何其他国家就GMP监管互认签署MRA,中国在GMP监管互认上的空白也成为中国药品出口的瓶颈所在。
但值得关注的是,近期PIC/S在官网上公布了2020年工作计划,其中特别提到,鉴于中国是世界药品市场原料药的主要供应国,PIC/S将持续加强与中国药监局的合作,以为中国药监局加入PIC/S打下基础。
03 美国监管机构对中国进口药品生产质量的监管
因尚未与美国、欧盟达成任何形式的GMP互认,就我国出口至美国、欧盟国家的药品以及我国药品生产企业受托生产的美国、欧盟上市药品,美国、欧盟国家的药品监管机构均有权按照美国、欧盟的GMP监管标准对位于我国境内的药品生产企业进行现场检查。
以美国为例,FDA对其有权检查的境外药品生产企业采用与其境内药品生产企业相同的cGMP监管标准,从cGMP现场检查的类型上看,也会分为常规监督检查、药品上市批准前检查和批准后检查及有因检查。在cGMP监管程序上,由FDA监管事务办公室(Office of Regulatory Affairs,ORA)负责具体执行对药品生产企业的现场检查职责,如果企业未满足cGMP的要求,检查官将以483表格的形式向生产企业明确现场检查缺陷,检查官也可能就483表格中记载的问题与企业的高管当面进行沟通,给予被查企业说明解释的机会。此外,FDA会向受检企业出具现场检查报告(Establishment Investigation Report,EIR),阐述检查结果。最终,生产企业会被识别为以下三种状态:不需要采取措施(No Action Indicated - NAI)、自愿采取措施(Voluntary Action Indicated - VAI)和官方需采取行动(Official Action Indicated - OAI)。对于被识别为OAI的生产企业或拒绝接受FDA监督检查的生产企业,FDA均可能签发警告信(Warning Letter)并予以公告,如果生产企业未能及时答复或补救效果未能令FDA满意,则对于中国的药品出口企业而言,将会影响到药品在美国的注册批准及进口;而对于中国的受托生产企业而言将可能丢失客户并影响其未来与其他美国药企的合作。对于FDA如何开展cGMP检查,我们在上一篇GMP比较法研究的文章《一文读懂中美欧GMP监管差异|GMP比较法研究(上)》中进行了更为详细的介绍,此处不再赘述。
虽然受限于监管资源,FDA无法对所有境外药品生产企业都进行检查,但FDA对亚洲地区,特别是对中国药企的关注度正逐年上升。特别是自FDA和欧盟达成MRA以来,双方节省了大量的监管资源并将更多的检查资源投入到其认定的产品质量与质量安全风险高的亚洲医药供应链地区。2016-2018年期间,FDA在印度和中国进行的药品境外检查最多,占总数的40%(见下表);[10]而在此期间,中国成为收到警告信数量最多的国家。[11]
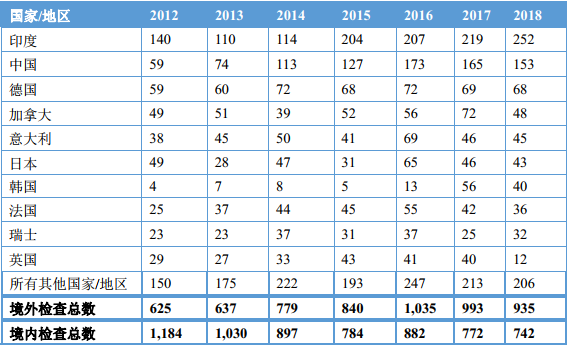
FDA开展境外检查情况[12]
为应对FDA日益增加的检查频率和日益严格的监管要求,我国制药企业还须时刻关注FDA的cGMP监管动态及监管要求的变化,积极完善生产质量管理体系。此外,针对FDA、EMA、WHO等境外药品监管/检查机构对境内生产企业开展的现场检查,我国国家药监局的核查中心也会组织对生产企业检查的现场观察,对于核查中心在现场观察或境外药品监管/检查机构出具的检查结果中发现的问题,也有可能促使核查中心后续开展独立的有因检查。
04 我国对进口药品及境外生产药品生产质量的监管
我国对进口药品的监管主要体现在以下几个方面:首先,进口至我国的药品应当依据《药品注册管理办法》的规定,向国家药监局药品审评中心(“审评中心”)申请,由审评中心对拟进口上市销售药品的安全性、有效性、质量可控性等进行审查,以获得《药品注册证书》。新《药品管理法》实施前,进口药品除了取得《新药证书》外,还需要取得《进口药品注册证》(针对从港澳台之外的地区进口的药品)或《医药产品注册证》(针对港澳台进口药品),目前均已合并为《药品注册证书》;但对于管制类药品(麻醉药品和国家规定范围内的精神药品),新《药品管理法》仍然保留了须取得国家药监局核发的《进口准许证》的要求。此外,进口药品还需在国家药监局与海关总署合作建立的药品和药材进口备案管理系统中完成备案,并填报报验单;海关凭药品监督管理部门出具的《进口药品通关单》放行;无《进口药品通关单》的,海关不予放行;此外,口岸所在地药品监督管理部门应当通知药品检验机构按照国务院药品监督管理部门的规定对进口药品进行抽查检验,允许药品进口的口岸由国务院药品监督管理部门会同海关总署提出,报国务院批准。
为确保进口药品质量,除了在新药注册环节对药品的安全性及和质量可控性进行把控外,国家药监局对药品境外生产企业的GMP现场检查也是对药品质量进行把控的重要环节,除进口药品外,境内药品上市许可持有人如果委托境外药品生产企业进行生产,该境外药品生产企业也需要根据新《药品管理法》及《药品生产监督管理办法》组织生产(包括符合GMP规定),并配合接受境外检查工作。
自2011年起,我国国家药监局会根据每年的整体部署对进口药品的境外生产企业进行现场检查工作。对于每年的整体部署,国家药监局都会发布《进口药品境外生产现场检查任务公告》,且会不时进行增补;选择检查对象的依据尚未有生效的法律文件明确规定,但通常是根据注册药品的品种特点、工艺特点、在中国的市场占有率,以及注册审评过程中的一些情况综合评估确定的。根据国家药监局于2017年8月发布的《药品境外检查管理规定(征求意见稿)》(“《征求意见稿》”)规定[13],国家药监局对拟检查品种及进口厂商,通过风险评估和随机抽查两种方式,制定年度检查计划并公开检查计划的基本信息,风险评估主要考虑药品的注册审评审批、日常监管、进口检验、不良反应监测以及投诉举报等风险信息,以确定年度检查计划。
从职责分工上,境外检查工作由国家药监局统筹安排,总局食品药品审核查验中心(“核查中心”)负责具体组织实施;中国食品药品检定研究院(“中检院”)、审评中心、总局药品评价中心(“评价中心”)等直属单位根据各自职能协助开展检查工作;上述单位除按规定提出药品境外检查任务外,中检院负责境外药品抽样的指导和检验工作;审评中心负责处于注册审评审批阶段品种的后续处理等工作;评价中心负责境外检查品种的相关评价工作。
此外,《征求意见稿》中还对检查流程进行了详细规定,具体流程图如下:

核查中心一旦发现药品存在质量问题或者其他安全隐患的,药品监督管理部门根据监督检查情况,将发出告诫信,并依据风险相应采取暂停生产、销售、使用、进口等控制措施。此外,境内药品上市许可持有人或进口药品的境外上市许可持有人或其境内代理还应当根据产品召回相关的法律规定于法定期限内对药品进行召回。
05 结语
药品GMP监管互认机制不仅能够帮助各国药品监管部门节省监管资源,更重要的是能够加快各国药品出口的步伐。在我国与他国建立起GMP监管互认体系之前,从事药品出口的企业还需时刻关注药品进口国对于GMP监管的要求和变化,以应对进口国药品监管机构随时可能发起GMP现场检查;而从事药品进口的进口厂商(包括药品境外上市许可持有人的境内代理)则应敦促境外生产企业持续遵守我国GMP规范,以免重蹈新基紫杉醇事件的覆辙。
[1] GMP|欧美深度手挽手,中国制药企业需要关注欧美GMP互认影响,CPhI制药在线
[2]李年苏,梁毅. PIC/S GMP 检查互信介绍及启示[J]. 中国现代应用药学, 2019, 36(14): 1833-1836
[3]李年苏,梁毅. PIC/S GMP 检查互信介绍及启示[J]. 中国现代应用药学, 2019, 36(14): 1833-1836
[4] PIC/S List of Pic/S Participating Authorities:https://picscheme.org/en/members
[5]李小春.无菌药品GMP检查国际合作项目试点工作正式启动[R] ,国际药品检查动态研究,2019年12月第4卷第6期
[6] FDA to Include Animal Drugs in EU MutualRecognition Agreement for Pharmaceutical Good Manufacturing Practice Inspections:https://www.fda.gov/animal-veterinary/cvm-updates/fda-include-animal-drugs-eu-mutual-recognition-agreement-pharmaceutical-good-manufacturing-practice
[7]张钟艺. FDA与EU的互认协议解析,沈药IFDPL国际药政通
[8] Annex to the Commission Decision on determiningthe Union position for a Decision of the Joint Committee set up under Article14 of the Agreement on Mutual Recognition between the European Community andthe United States of America, in order to amend the Sectoral Annex on GMP: http://trade.ec.europa.eu/doclib/docs/2017/february/tradoc_155398.pdf
[9] Mutual Recognition Agreement (MRA): https://www.fda.gov/international-programs/international-arrangements/mutual-recognition-agreement-mra
[10]李小春. FDA药品境外检查工作情况介绍[R] ,国际药品检查动态研究,2019年12月第4卷第6期
[11]国际药政通,2018财年FDA药品GMP警告信分析[R] ,国际药品检查动态研究,2019年4月第4卷第2期
[12]图片来自:李小春. FDA药品境外检查工作情况介绍[R] ,国际药品检查动态研究,2019年12月第4卷第6期
[13]《药品境外检查管理规定(征求意见稿)》已于2017年9月24日完成了征求意见的工作,但因尚未生效实施,本文中关于《药品境外检查管理规定(征求意见稿)》规定的内容仅供参考。
在上一篇关于药品GMP比较法研究的文章《一文读懂中美欧GMP监管差异|GMP比较法研究(上)》中,我们从监管法律体系、监管机构及人员以及GMP检查程序三个方面对我国与美国、欧盟的药品GMP监管进行了比较。正因为各国监管机构的GMP监管标准及程序存在差别,在涉及药品进出口及跨境委托生产时,如何就该等药品开展GMP监管成为各国药品监管棘手的难题,也成为开展药品进出口及跨境委托、受托生产业务的药品企业最为关心的问题之一。前段时间,我国国家药监局叫停美国Celgene Corporation注射用紫杉醇(白蛋白结合型)进口并取消其集中采购中选资格,就是国家药监局在执行对从境外进口药品的监管职能,其依据为我国的GMP监管标准。
本文将从国际上通行的GMP监管互认制度出发,介绍美国、欧盟及我国目前的药品GMP监管互认状态,并基于此谈谈美国FDA和我国国家药监局是如何开展GMP跨国监管的。
01 药品GMP监管互认概述
基于对药品质量的把控及监管要求,各国的药品监管机构通常会对于以下两类境外生产药品(“境外生产药品”)的生产企业按照本国的GMP标准进行现场检查:①进口药品的生产企业,以及②境内药品的境外受托生产企业,并根据检查结果决定是否允许受检药品进口、上市或销售,从而保证本国公民的用药安全。但因跨境监管耗时耗力,有限的监管资源越来越难满足各国对境外生产药品的跨境检查需求,在各国家/地域的药品监管机构之间实行GMP监管互认,能够在保证药品安全的同时促进合规标准的一致性发展,避免了不同国家/地域的药品监管机构对药品生产的重复性检查工作,节省了监管资源,同时也降低了药企应对重复性检查的成本。
药品GMP监管互认存在以下几种主要方式:
1.1 签署互认协议
各国家/地域的药品监管机构基于对其他国家/地域药品监管能力的评估和认可签署互认协议(Mutual Recognition Agreements,“MRA”),并就互认范围、适用于现场检查豁免或必须进行现场检查的品种,以及互认程序、信息共享、药物警戒机制等双方关注的问题予以约定,以实现一方监管机构对另一方监管机构GMP检查结果的承认。但是,MRA的签署并不意味着一方监管机构一定不会重复对方监管机构已执行过的检查,签署双方的监管机构通常都留有在例外情况下对另一方管辖范围内的药品生产企业执行GMP检查的权利。[1]
1.2 加入PIC/S
国际药品监查合作计划(The Pharmaceutical Inspection Co-operation Scheme, “PIC/S”)成立于1995年11月,是世界上唯一由各国GMP检查权责机关组成的国际合作组织,其成立的目的在于促进GMP的国际合作及标准一致化。PIC/S是一个国际药品生产GMP互认组织,各成员国在自愿的基础上相互承认官方GMP检查;与MRA不同的是,PIC/S搭建的是一个非约束性、非正式的合作平台[2],而MRA对于签署双方具有法律约束力。该组织的任务包括:促进各国卫生主管机关的检查互信,制定统一的GMP规范,统一GMP认证系统,对检查认证人员统一培训(如即将于2020年11月在泰国曼谷举办的“如何成为优秀的GMP检查员”研讨会),促进GMP信息的交流。
为此,PIC/S制定了GMP检查互信指南,概述了境外药品生产企业GMP合规性的非现场评估过程,并确定在何时可通过其他或当地监管机构的监管动作来确认并保证企业处于可接受的GMP合规水平范围内,从而替代现场检查,避免了监管机构间的重复工作。[3]
PIC/S目前共有50余个监管机构成员,遍布澳大利亚、加拿大、捷克、丹麦、芬兰、法国、德国、新加坡、日本、英国、美国、台湾和香港等国家/地区。[4]监管机构成员均需要满足并维持PIC/S GMP标准,以作为其GMP检查互认的基础,该组织颁发的GMP证书在PIC/S组织成员之间可以相互认可,所以加入PIC/S是进入国际市场的快捷通道之一。
1.3 其他合作项目
除上述两种方式外,各国家、国际组织间还会不定期就特定项目开展合作,如2008-2016年的原料药检查国际合作项目,以及2019年12月刚刚启动且将持续至少2年时间的无菌药品GMP检查国际合作项目(“无菌药品合作项目”),参与方有FDA、EMA、法国药品保健品署(ANSM)、英国药品和健康产品管理局(MHRA)、澳大利亚药品管理局(TGA)、加拿大卫生部(HC)、日本药品医疗器械综合机构(PMDA)及世界卫生组织(WHO)。无菌药品合作项目的范围为人用化学无菌药品,以及某些治疗性生物制品(如单克隆抗体、重组蛋白),不含疫苗、细胞和基因疗法以及血液制品;根据项目工作计划,参与机构将协调和共享检查计划,并鼓励各方根据各自确定的GMP规范开展联合检查(避免独立或同步检查),每月共享检查结果。[5]
02 药品GMP监管的互认现状
2.1 欧盟对其他国家/地区GMP监管的承认
截至目前,欧盟已分别与澳大利亚、加拿大、以色列、日本、新西兰、瑞士及美国共7个国家签署了MRA,以在欧盟与该等国家之间实现药品GMP监管互认,以及就监管及质量问题进行信息共享等目的,但欧盟与各国签署的MRA范围均不相同。
在欧盟已签署的MRA中,最引人关注的要数与美国FDA签署的MRA。
这份MRA项下的互认范围为:
①双方在其领土范围内对已上市产品执行的生产检查;
②双方在其领土范围内对产品执行的上市前生产检查;
③双方在其领土范围外对产品执行的生产检查。
所覆盖的产品类型包括人用药品和生物制品(具体产品类别可在MRA的附录中找到);此外,欧盟和FDA应于2017年12月15日前决定是否将兽药包含在MRA所覆盖的产品类型中(根据FDA于2020年3月13日发布的公告,FDA兽药中心已于2019年12月向EC报告将兽药作为MRA所覆盖的产品,且欧盟已基本同意[6]),并于2022年7月15日前决定是否将人用疫苗和源于血浆的产品包含在MRA所覆盖的产品类型中;人血、人类血浆、人类组织和器官,以及兽用免疫物质不在MRA所覆盖的产品类型中。此外,值得关注的是,欧盟和FDA的MRA中也约定了一方拒绝认可另一方GMP检查结果的特殊情形,即在检查报告中存在原料不符或不足的迹象,在上市后监管中发现产品存在质量缺陷,或存在与产品质量或消费者安全相关的其他特殊证据等特殊情况下,欧盟和FDA可选择不接受对方就其领土范围内的生产企业所发布的官方GMP文件。[7][8]
欧盟负责MRA起草和谈判工作的是欧盟委员会(EC),EMA则负责向EC提供咨询服务,并在MRA签署后负责后续的执行工作,包括作为互认监管机构与欧盟成员国监管信息的对接口,负责就已签署的MRA进行答疑和管理,管理数据库,并与成员国的监管机构进行数据共享等。
2.2 美国对其他国家/地区GMP监管的承认
2012年7月9日,美国国会通过《食品、药品管理安全与创新法案》(Food and Drug Administration Safety and Innovation Act),其中规定,如果FDA认为其他国家/地区的监管机构能够达到美国监管标准,则FDA有权与该境外监管机构对该监管机构境内药品生产的监管进行互认。[9]但美国的药品GMP互认门槛很高且FDA对互认非常谨慎,根据FDA官网显示,美国截至目前仅与欧盟签署了MRA,且FDA虽然在2017年7月与欧盟签署了MRA,但因对欧盟各成员国GMP监管机构的检查工作质量持保守态度,因此并没有在MRA生效时就对所有欧盟成员国的GMP监管进行承认,而是约定了过渡期,并在过渡期内比照美国的现场审查标准对欧盟成员国的生产检查稳定性逐个进行审查,历时两年的时间才完成了对欧盟28个国家GMP监管的承认。
2.3 我国与其他国家/地区GMP监管体系的互认
2017年7月,国际人用药品注册技术协调会(ICH)确认中国药监局成为ICH第8个监管机构成员,使得中国药品在研发和注册领域的国际化道路上迈出了重要一步,但在药品生产监管领域,中国的监管体系还未加入PIC/S,中国也尚未与任何其他国家就GMP监管互认签署MRA,中国在GMP监管互认上的空白也成为中国药品出口的瓶颈所在。
但值得关注的是,近期PIC/S在官网上公布了2020年工作计划,其中特别提到,鉴于中国是世界药品市场原料药的主要供应国,PIC/S将持续加强与中国药监局的合作,以为中国药监局加入PIC/S打下基础。
03 美国监管机构对中国进口药品生产质量的监管
因尚未与美国、欧盟达成任何形式的GMP互认,就我国出口至美国、欧盟国家的药品以及我国药品生产企业受托生产的美国、欧盟上市药品,美国、欧盟国家的药品监管机构均有权按照美国、欧盟的GMP监管标准对位于我国境内的药品生产企业进行现场检查。
以美国为例,FDA对其有权检查的境外药品生产企业采用与其境内药品生产企业相同的cGMP监管标准,从cGMP现场检查的类型上看,也会分为常规监督检查、药品上市批准前检查和批准后检查及有因检查。在cGMP监管程序上,由FDA监管事务办公室(Office of Regulatory Affairs,ORA)负责具体执行对药品生产企业的现场检查职责,如果企业未满足cGMP的要求,检查官将以483表格的形式向生产企业明确现场检查缺陷,检查官也可能就483表格中记载的问题与企业的高管当面进行沟通,给予被查企业说明解释的机会。此外,FDA会向受检企业出具现场检查报告(Establishment Investigation Report,EIR),阐述检查结果。最终,生产企业会被识别为以下三种状态:不需要采取措施(No Action Indicated - NAI)、自愿采取措施(Voluntary Action Indicated - VAI)和官方需采取行动(Official Action Indicated - OAI)。对于被识别为OAI的生产企业或拒绝接受FDA监督检查的生产企业,FDA均可能签发警告信(Warning Letter)并予以公告,如果生产企业未能及时答复或补救效果未能令FDA满意,则对于中国的药品出口企业而言,将会影响到药品在美国的注册批准及进口;而对于中国的受托生产企业而言将可能丢失客户并影响其未来与其他美国药企的合作。对于FDA如何开展cGMP检查,我们在上一篇GMP比较法研究的文章《一文读懂中美欧GMP监管差异|GMP比较法研究(上)》中进行了更为详细的介绍,此处不再赘述。
虽然受限于监管资源,FDA无法对所有境外药品生产企业都进行检查,但FDA对亚洲地区,特别是对中国药企的关注度正逐年上升。特别是自FDA和欧盟达成MRA以来,双方节省了大量的监管资源并将更多的检查资源投入到其认定的产品质量与质量安全风险高的亚洲医药供应链地区。2016-2018年期间,FDA在印度和中国进行的药品境外检查最多,占总数的40%(见下表);[10]而在此期间,中国成为收到警告信数量最多的国家。[11]
FDA开展境外检查情况[12]
为应对FDA日益增加的检查频率和日益严格的监管要求,我国制药企业还须时刻关注FDA的cGMP监管动态及监管要求的变化,积极完善生产质量管理体系。此外,针对FDA、EMA、WHO等境外药品监管/检查机构对境内生产企业开展的现场检查,我国国家药监局的核查中心也会组织对生产企业检查的现场观察,对于核查中心在现场观察或境外药品监管/检查机构出具的检查结果中发现的问题,也有可能促使核查中心后续开展独立的有因检查。
04 我国对进口药品及境外生产药品生产质量的监管
我国对进口药品的监管主要体现在以下几个方面:首先,进口至我国的药品应当依据《药品注册管理办法》的规定,向国家药监局药品审评中心(“审评中心”)申请,由审评中心对拟进口上市销售药品的安全性、有效性、质量可控性等进行审查,以获得《药品注册证书》。新《药品管理法》实施前,进口药品除了取得《新药证书》外,还需要取得《进口药品注册证》(针对从港澳台之外的地区进口的药品)或《医药产品注册证》(针对港澳台进口药品),目前均已合并为《药品注册证书》;但对于管制类药品(麻醉药品和国家规定范围内的精神药品),新《药品管理法》仍然保留了须取得国家药监局核发的《进口准许证》的要求。此外,进口药品还需在国家药监局与海关总署合作建立的药品和药材进口备案管理系统中完成备案,并填报报验单;海关凭药品监督管理部门出具的《进口药品通关单》放行;无《进口药品通关单》的,海关不予放行;此外,口岸所在地药品监督管理部门应当通知药品检验机构按照国务院药品监督管理部门的规定对进口药品进行抽查检验,允许药品进口的口岸由国务院药品监督管理部门会同海关总署提出,报国务院批准。
为确保进口药品质量,除了在新药注册环节对药品的安全性及和质量可控性进行把控外,国家药监局对药品境外生产企业的GMP现场检查也是对药品质量进行把控的重要环节,除进口药品外,境内药品上市许可持有人如果委托境外药品生产企业进行生产,该境外药品生产企业也需要根据新《药品管理法》及《药品生产监督管理办法》组织生产(包括符合GMP规定),并配合接受境外检查工作。
自2011年起,我国国家药监局会根据每年的整体部署对进口药品的境外生产企业进行现场检查工作。对于每年的整体部署,国家药监局都会发布《进口药品境外生产现场检查任务公告》,且会不时进行增补;选择检查对象的依据尚未有生效的法律文件明确规定,但通常是根据注册药品的品种特点、工艺特点、在中国的市场占有率,以及注册审评过程中的一些情况综合评估确定的。根据国家药监局于2017年8月发布的《药品境外检查管理规定(征求意见稿)》(“《征求意见稿》”)规定[13],国家药监局对拟检查品种及进口厂商,通过风险评估和随机抽查两种方式,制定年度检查计划并公开检查计划的基本信息,风险评估主要考虑药品的注册审评审批、日常监管、进口检验、不良反应监测以及投诉举报等风险信息,以确定年度检查计划。
从职责分工上,境外检查工作由国家药监局统筹安排,总局食品药品审核查验中心(“核查中心”)负责具体组织实施;中国食品药品检定研究院(“中检院”)、审评中心、总局药品评价中心(“评价中心”)等直属单位根据各自职能协助开展检查工作;上述单位除按规定提出药品境外检查任务外,中检院负责境外药品抽样的指导和检验工作;审评中心负责处于注册审评审批阶段品种的后续处理等工作;评价中心负责境外检查品种的相关评价工作。
此外,《征求意见稿》中还对检查流程进行了详细规定,具体流程图如下:
核查中心一旦发现药品存在质量问题或者其他安全隐患的,药品监督管理部门根据监督检查情况,将发出告诫信,并依据风险相应采取暂停生产、销售、使用、进口等控制措施。此外,境内药品上市许可持有人或进口药品的境外上市许可持有人或其境内代理还应当根据产品召回相关的法律规定于法定期限内对药品进行召回。
05 结语
药品GMP监管互认机制不仅能够帮助各国药品监管部门节省监管资源,更重要的是能够加快各国药品出口的步伐。在我国与他国建立起GMP监管互认体系之前,从事药品出口的企业还需时刻关注药品进口国对于GMP监管的要求和变化,以应对进口国药品监管机构随时可能发起GMP现场检查;而从事药品进口的进口厂商(包括药品境外上市许可持有人的境内代理)则应敦促境外生产企业持续遵守我国GMP规范,以免重蹈新基紫杉醇事件的覆辙。
[1] GMP|欧美深度手挽手,中国制药企业需要关注欧美GMP互认影响,CPhI制药在线
[2]李年苏,梁毅. PIC/S GMP 检查互信介绍及启示[J]. 中国现代应用药学, 2019, 36(14): 1833-1836
[3]李年苏,梁毅. PIC/S GMP 检查互信介绍及启示[J]. 中国现代应用药学, 2019, 36(14): 1833-1836
[4] PIC/S List of Pic/S Participating Authorities:https://picscheme.org/en/members
[5]李小春.无菌药品GMP检查国际合作项目试点工作正式启动[R] ,国际药品检查动态研究,2019年12月第4卷第6期
[6] FDA to Include Animal Drugs in EU MutualRecognition Agreement for Pharmaceutical Good Manufacturing Practice Inspections:https://www.fda.gov/animal-veterinary/cvm-updates/fda-include-animal-drugs-eu-mutual-recognition-agreement-pharmaceutical-good-manufacturing-practice
[7]张钟艺. FDA与EU的互认协议解析,沈药IFDPL国际药政通
[8] Annex to the Commission Decision on determiningthe Union position for a Decision of the Joint Committee set up under Article14 of the Agreement on Mutual Recognition between the European Community andthe United States of America, in order to amend the Sectoral Annex on GMP: http://trade.ec.europa.eu/doclib/docs/2017/february/tradoc_155398.pdf
[9] Mutual Recognition Agreement (MRA): https://www.fda.gov/international-programs/international-arrangements/mutual-recognition-agreement-mra
[10]李小春. FDA药品境外检查工作情况介绍[R] ,国际药品检查动态研究,2019年12月第4卷第6期
[11]国际药政通,2018财年FDA药品GMP警告信分析[R] ,国际药品检查动态研究,2019年4月第4卷第2期
[12]图片来自:李小春. FDA药品境外检查工作情况介绍[R] ,国际药品检查动态研究,2019年12月第4卷第6期
[13]《药品境外检查管理规定(征求意见稿)》已于2017年9月24日完成了征求意见的工作,但因尚未生效实施,本文中关于《药品境外检查管理规定(征求意见稿)》规定的内容仅供参考。